
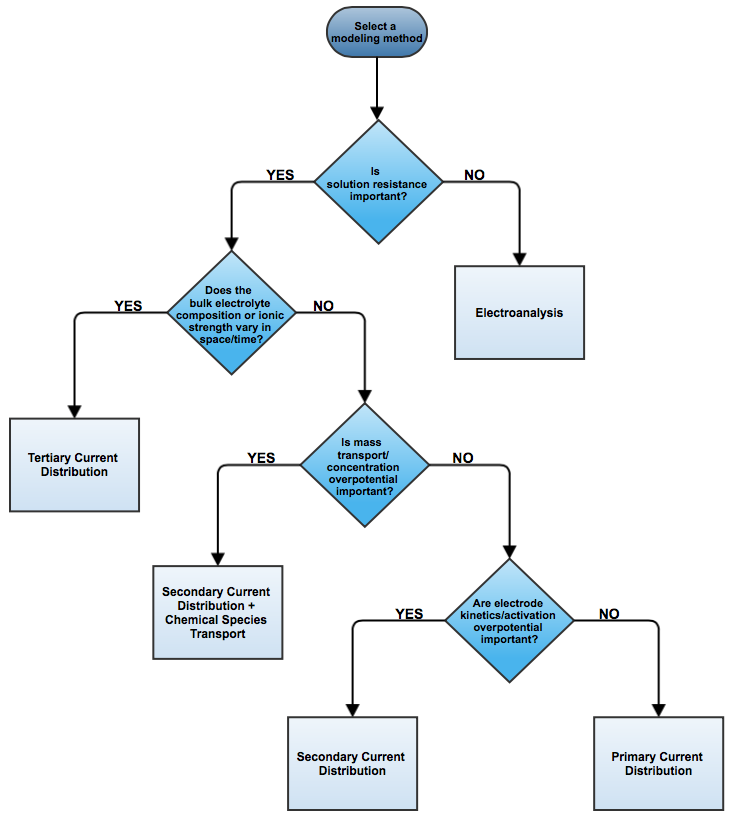
When designing electrochemical cells, we consider the three classes of current distribution in the electrolyte and electrodes: primary, secondary, and tertiary. We recently introduced the essential theory of current distribution. Here, we illustrate the different current distributions with a wire electrode example to help you choose between the current distribution interfaces in COMSOL Multiphysics for your electrochemical cell simulation.
Introducing the Three Current Distribution Interfaces
As you saw in the previous blog post, we can use an example model of a wire electrode to compare the three current distribution interfaces. Here is the geometry again:
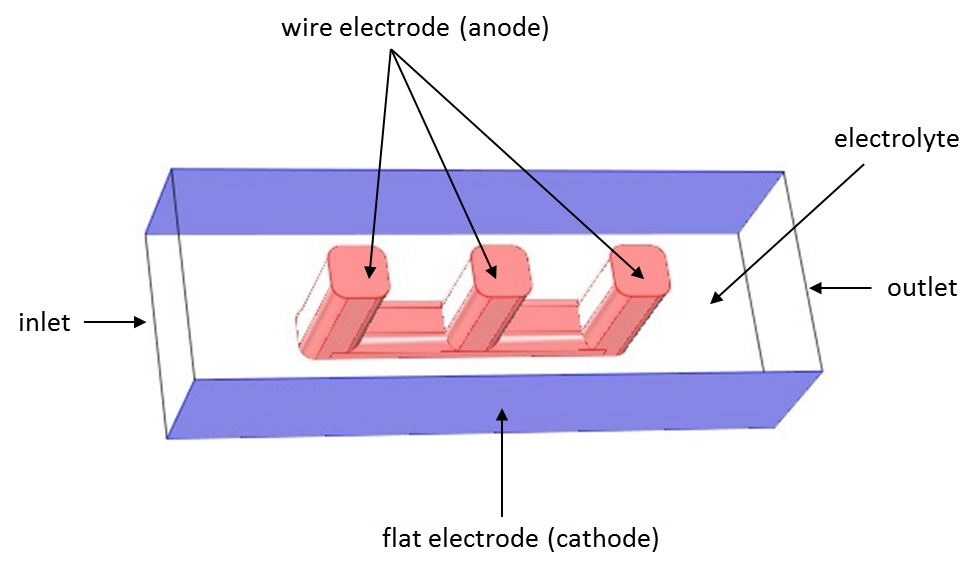
Wire electrode model solved using COMSOL Multiphysics. Electrolyte is allowed to flow in the open volume between the wire and flat surfaces.
The same geometry is considered in all three cases presented here: a wire electrode structure is placed between two flat electrode surfaces. The electrochemical cell can be seen as a unit cell of a larger wire-mesh electrode, which is an electrochemical cell set-up common in many large-scale industrial processes.
Recap of the Essential Equations
Below are the essential equations we mentioned in detail previously:
Nernst-Planck equation:
(1)
Current density expression with the Nernst-Planck equation:
(2)
General electrolyte current conservation:
Primary Current Distribution
The primary current distribution accounts only for losses due to solution resistance, neglecting electrode kinetic and concentration-dependent effects. The charge transfer in the electrolyte is assumed to obey Ohm’s law. We are making two assumptions here: first, that the electrolyte is electroneutral, which cancels out the convective contribution to the current density in equation (2), and second, that composition variations in the electrolyte are negligible (it is homogeneous), which cancels out the diffusive contribution to the current density in equation (2) and allows us to treat the ionic strength as a constant. Hence, the remaining term of equation (2) results in Ohm’s law for electrolyte current density.
At the electrode-electrolyte interface we assume that the electrolysis reaction is so fast that we can neglect the influence of electrode kinetics, and so the potential difference at the electrode-electrolyte boundary deviates negligibly from its equilibrium value. In other words, there is no activation overpotential and an arbitrary current density can occur through electrolysis. Therefore, the primary current distribution only depends on the geometry of the anode and cathode.
The Primary Current Distribution interface in COMSOL Multiphysics defines two dependent variables: one for the electric potential in the electrolyte (\phi_l\) and another for the electric potential in the electrodes (\phi_s\). With the above described assumptions for a primary current distribution, you get the following equations to be solved:
Electrode: \textbf{i}_s = -\sigma_s\nabla\phi_s\ with \nabla\cdot\textbf{i}_s = Q_s
Electrolyte: \textbf{i}_l = -\sigma_l\nabla \phi_l\ with \nabla\cdot\textbf{i}_l = Q_l
Electrode-Electrolyte-Interface: \phi_s-\phi_l = E_{\mathrm{eq},m}
Here, \sigma_l denotes the conductivity of the electrolyte, which is constant by the above assumptions. The index s represents the electrode and l the electrolyte. E_{\mathrm{eq},m} denotes the equilibrium potential for reaction m.
In the following picture, we show the primary current density distribution for our wire electrode example. As you can see, the current density distribution is highest at the corners of the wires directly facing the cathode plates, and close to zero at the central parts of the wire structure that are geometrically shielded from the cathode.
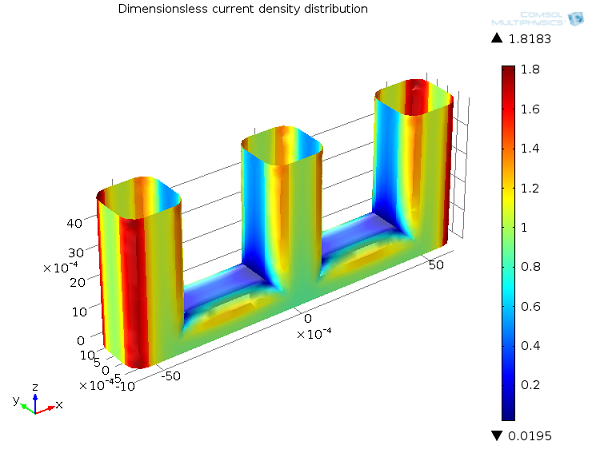
Primary current distribution, Ecell = 1.65 V. Current density distribution on the anode (dimensionless).
When Do I Use the Primary Current Distribution Interface?
You can use this class of current distribution for modeling cells where you have a relatively high electrolyte concentration (in relation to current density) or vigorous mixing in the electrolyte, allowing the assumption of a uniform electrolyte concentration. In addition, the electrochemical reactions have to be fast enough for negligible resistance to be associated with the reaction, compared to the magnitude of the ohmic losses (solution resistance).
One application of these conditions is at the anodes in systems for imposed current cathodic protection, while a constant current corresponding to the mass transport-limited current for oxygen reduction can be set as a boundary condition at the cathode in the primary current density interface (here’s an example of this). This can also be a valuable approximation for electrochemical processes involving relatively fast reactions, such as the oxidation of chloride ions in the chlor-alkali process.
Since the Primary Current Distribution interface is easy to solve and involves no nonlinear kinetic expressions, it is often suitable to use in order to calculate a baseline approximation before approaching a more complex model.
Secondary Current Distribution
The secondary current distribution accounts for the effect of the electrode kinetics in addition to solution resistance. The assumptions about the electrolyte composition and behavior are the same as for the primary current distribution, resulting in Ohm’s law for electrolyte current. The difference between the primary and secondary current distributions lies in the description of the electrochemical reaction at the interface between an electrolyte and an electrode.
Here, the influence of electrode kinetics is included; the potential difference may differ from its equilibrium value due to additional impedance associated with the finite rate of the electrolysis reaction. The difference between the actual potential difference and the equilibrium potential difference is the activation overpotential (\eta). Thus, you get the same domain equations as in the Primary Current Distribution interface, but the electrode-electrolyte interface equation differs according to the overpotential:
Electrode: \textbf{i}_s = -\sigma_s\nabla\phi_s\ with \nabla\cdot\textbf{i}_s = Q_s
Electrolyte: \textbf{i}_l = -\sigma_l\nabla \phi_l\ with \nabla\cdot\textbf{i}_l = Q_l
Electrode-Electrolyte-Interface: \eta_m = \phi_s-\phi_l-E_{eq,m}
In the Secondary Current Distribution interface, the current density due to the electrochemical reactions is described as a function of the overpotential. The physics interface can use any relation between current density and overpotential, with common examples such as the Butler-Volmer equation (3) and the Tafel equation included as built-in options.
(3)
In the Butler-Volmer equation above, for reaction m:i_{loc,m} denotes the local charge transfer current density, i_{0,m} the exchange current density, and \alpha_{a,m} the anodic and \alpha_{c,m} the cathodic charge transfer coefficient. R is the universal gas constant. This equation describes the case when the charge transfer of one electron is the rate determining step in the net charge transfer reaction. The expression can be derived by analogy to the Arrhenius equation for a homogeneous chemical reaction, by assuming the free energy of the charged species to be influenced by the potential. Hence, the activation energy changes with the potential difference at the electrode-electrolyte interface.
The sum of all electrode reaction currents is implemented as a current density condition on the boundary between an electrode and an electrolyte domain according to:
The additional capacitive current i_\mathrm{DL} arises from charge and discharge of the electrical double layer.
In general, accounting for the effect of electrode kinetics by means of an activation overpotential will tend to make the current distribution more uniform. You can see this in the wire electrode example in the figure below.
Compared to the primary current distribution, the secondary current distribution is smoother, with a smaller difference between the minimum and maximum values. When the activation overpotential is included, a high local current density would introduce a high local activation overpotential at the electrode surface, which causes the current to naturally take a different path. To look at this another way, you can understand the electrochemical reaction as proceeding at a finite rate. In some regions, the reaction is kinetically limited, and so the distribution of current densities over the surface is less extreme than in the case when the reaction can proceed arbitrarily quickly.
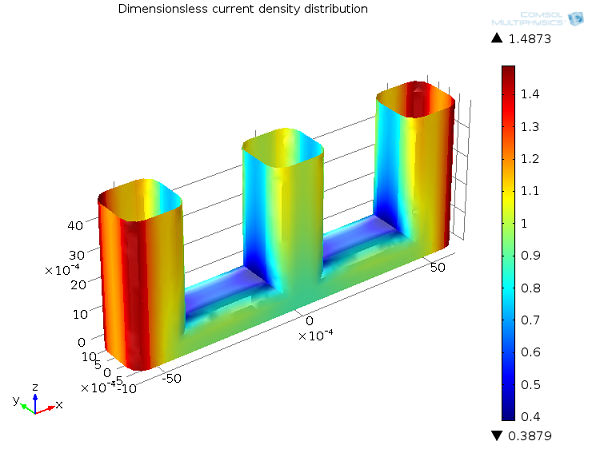
Secondary current distribution, Ecell = 1.65 V. Current density distribution on the anode (dimensionless).
When Do I Use the Secondary Current Distribution Interface?
Secondary Current Distribution is the workhorse interface for modeling industrial applications in electrochemistry. You can use this class of current distribution for modeling cells where you can neglect concentration overpotential, due to good mixing or relatively high electrolyte concentration, but when the electrode kinetics cause losses that are not negligible compared to the ohmic losses. In industrial applications it is usually not a problem to provide an electrolyte of high concentration with vigorous mixing. You can also use the Secondary Current Distribution interface as a first step in your simulation of electrochemical cells to estimate the activation losses, before you eventually introduce concentration-dependent reaction kinetics.
Tertiary Current Distribution
The tertiary current distribution accounts for the effect of variations in electrolyte composition and ionic strength on the electrochemical process, as well as solution resistance and electrode kinetics. To do this, it solves the Nernst-Planck equation (1) explicitly for each chemical species to describe its mass transport through diffusion, migration, and convection. Additionally, the species concentrations are subject to the electroneutrality approximation. The kinetic expressions for the electrochemical reactions account for both activation and concentration overpotential, meaning that the rate of an electrolysis reaction can be transport-limited by exhaustion of the reactant at the electrode-electrolyte interface. This implies that all ions and all electroactive species in the electrolyte must be included in the model.
Unlike the primary and secondary current distributions, the electrolyte current density is no longer assumed to follow Ohm’s law in the tertiary current distribution. The imposition of electroneutrality still means that convective flux does not contribute to the current density, due to equation (2), but now the influence of the concentration variations in the electrolyte cannot be neglected. Therefore, the diffusion term in equation (2) may be nonzero.
At the electrode-electrolyte interface, the current density of charge transfer reactions is expressed as a function not only of the overpotential, but also of the concentration of the electroactive species at the interface. For a reaction rate determined by a one-electron charge transfer step, the reaction kinetics is expressed using a Butler-Volmer expression for the charge transfer current density i_{loc,m} (compare with equation (3)), which in this case can contain concentration dependencies.
The Tertiary Current Distribution interface in the COMSOL software solves for the electrolyte potential (\phi_l\), the electrode potential (\phi_s\), and the set of species concentrations c_i. With the assumptions described above you get the following equations:
Electrode: \textbf{i}_s = -\sigma_s\nabla\phi_s\ with \nabla\cdot\textbf{i}_s = Q_s
Electrolyte: \textbf{i}_l = F\sum_{i=1}^n z_i (-D_i\nabla c_i-z_i u_{m,i} F c_i\nabla \phi_l) with \nabla\cdot\textbf{i}_l = Q_l
Electrolyte electroneutrality: \sum_i z_ic_i = 0
Electrode-Electrolyte-Interface: \eta_m = \phi_s-\phi_l-E_{eq,m}
Typical current density expression: i_{loc,m} = i_{0,m}\left(\frac{c_\mathrm{Red}}{c_\mathrm{ref}} e^\frac{\alpha_{a,m} F \eta_m}{RT}-\frac{c_\mathrm{Ox}}{c_\mathrm{ref}}e^\frac{-\alpha_{c,m} F \eta_m }{RT}\right)
It is essential that the reference concentration c_\mathrm{ref} is the same for all species involved in a reaction. This ensures that at zero current density (equilibrium) the overpotential obeys the thermodynamic Nernst equation.
In the image below, you can see the tertiary current distribution for the wire example. Due to the dependence of the concentration, the tertiary current distribution becomes influenced by the flow of the electrolyte and hence the availability of the reactant by mass transport. Where the flow velocity is small between the wires, electrolyte consumed to draw Faradaic current is not replenished, leading to a depletion zone of the reactant in these parts in the cell. This significantly lowers the local current density, which can be described as “mass-transport limited”, leading a greater amount of the current to be drawn from the outer edges of the wires. A corresponding increased voltage drop is observed due to the transport limitation of current: this is the “concentration overpotential”.
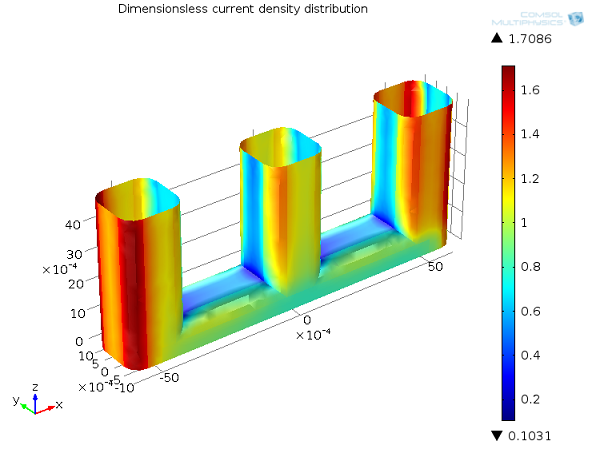
Tertiary current distribution, Ecell = 1.65 V. Current density distribution on the anode (dimensionless).
When Do I Use the Tertiary Current Distribution Interface?
You can use this class of current distribution for modeling cells with poor mixing or relatively low electrolyte concentration (compared to net current density), such that the electrolyte composition varies significantly throughout the cell and the resistive losses cannot be described by Ohm’s law. Solving the Nernst-Planck equations for all species concentrations with concentration of current and electroneutrality makes the equation set nonlinear and very complicated for the tertiary current distribution, which results in more time and memory storage requirements for the simulation. It is good practice to predict and understand the likely behavior of an electrochemical cell with secondary current distribution before modeling the tertiary current distribution.
What Other Options Are There?
The primary, secondary, and tertiary current distributions distinguish successive levels of approximation in the analysis of the current-voltage relation of an electrochemical cell. There are other modeling approaches that may be suitable, however, to extract maximum information about a cell’s behavior while minimizing the complexity of the model as much as possible.
Secondary Current Distribution with Chemical Species Transport
In cases where the current density may be limited by mass transport of the electroactive species, but the electrolyte composition remains near-constant, it may not be necessary to solve for the full tertiary current distribution. Instead, the constant ionic strength means that we can assume that the solution obeys Ohm’s law with a constant conductivity, and so Secondary Current Distribution is used to solve for the electrolyte potential. However, the kinetic rate law is made concentration-dependent by coupling to a chemical species transport model that solves for the diffusion of the chemical species (and, where necessary, their migration and convection).
In fact, this is the method used for the tertiary current distribution of the wire electrode example, since it is the depletion of the reactant rather than the bulk electrolyte species that has the dominating effect. You can see another example of this coupling in the orange battery model. Also, this partial coupling of charge transport with mass transport is a very common approach in the analysis of batteries and fuel cells.
Electroanalysis
A special case of the above occurs when the inert (supporting) electrolyte is in considerable excess compared to the quantity of reacting (electroactive) species. Hence, the ionic strength of the solution is large compared to the Faradaic current density. In this case, the electric field is small and so the electrolyte potential is almost constant — solution resistance does not contribute noticeably to the behavior of the electrochemical cell.
In cases where solution resistance is unimportant, but electrode kinetics (activation) and mass transport of the electroactive species are important, you can use the Electroanalysis interface. This is a chemical species transport interface solving the diffusion-convection equation for mass transport, which incorporates electrode kinetic boundary conditions to drive a flux of the chemical species at electrode-electrolyte interfaces as a function of the local overpotential.
The electroanalytical approximation of zero solution resistance applies to the standard experimental set-ups for electrochemical techniques such as cyclic voltammetry, chronoamperometry, and electrochemical impedance spectroscopy. You can see an example of a cyclic voltammetry model using this approximation in our Model Gallery.
Concluding Thoughts
This blog post has discussed the three current distribution interfaces available in the four electrochemical add-on modules for COMSOL Multiphysics, and when and why you should use each of them. The strength of the COMSOL Multiphysics software is that it offers you the ability to model all classes of current distributions (primary, secondary, and tertiary), and therefore provides you with the flexibility to gradually introduce and control the complexity of the theoretical model used to analyze an electrochemical cell.
If you are interested in using COMSOL Multiphysics for your electrochemical cell design, or have a question that isn’t addressed here, please contact us.




Comments (10)
Lasse Murtomäki
October 29, 2014Hi
Although I am an electrochemist, I cannot quite understand the implementation of the tertiary current distribution in Comsol. The current-overpotential equation is usually written as
i/i0 = (C_R/C_R,bulk)*exp(a*n*f*eta) – (C_O/C_O,bulk)*exp((a-1)*n*f*eta)
Above you write “It is essential that the reference concentration is the same for all species involved in a reaction. This ensures that at zero current density (equilibrium) the overpotential obeys the thermodynamic Nernst equation”. At zero current overpotential is zero, and the surface concentrations are equal to bulk ones. Hence, I do not get this formulation or statement.
In the wire electrode example, concentration polarizarion is taken into account via a separate “Transport of dilutes species” node which is then coupled at the electrode surface with the local current density. Is it thus possible to do this with a single “Tertiary current distribution” node instead, using concentration dependent kinetics in the electrode reaction? I have not succeeded in doing this with a simple 1D model either. Either the model does not nothing or cannot find initial values.
Best regards
Lasse Murtomäki
Edmund Dickinson
October 30, 2014Lasse:
It is fine to use different bulk concentrations, as in the equation you write. However, if these are different for the two species of the redox couple, then in this case you must make sure that the “Equilibrium potential” against which the overpotential is measured is corrected (manually) from the formal electrode potential, to account for the unequal concentrations according to the Nernst equation. If you use a common reference concentration as we suggest here, then in the model you can define the equilibrium potential as the formal electrode potential directly. We recommend the latter method because there is less possibility for mistake.
The advantage of Secondary Current Distribution + Transport of Diluted Species over Tertiary Current Distribution, Nernst-Planck is that the former method usually gives a much more linear equation set. Only mass transport of diluted reactants is considered, rather than mass transport of all species including inert electrolyte. If you need advice on setting up a simple model, you can contact us at http://www.comsol.com/support/.
Edmund Dickinson
COMSOL UK
Tim Holme
August 10, 2017Where do you select the current distribution interface (primary vs secondary vs tertiary)? I’m using the Li-ion battery module and I can’t find this option.
Thank you.
Melanie Pfaffe
August 11, 2017Dear Tim:
The current distribution interfaces are available within the Batteries and Fuel Cells Module. They are general interfaces which are not predefined for a specific appliation. For your case of Lithium-ion batteries we have the predefined interface called Lithium-Ion Battery intrface, which already includes the needed functionality of the current distribution interfaces. The Lithium-Ion Battery interface is used to compute the potential and current distributions in a lithium-ion battery. Multiple intercalating electrode materials can be used, and voltage losses due to solid-electrolyte-interface (SEI) layers are also included.
Yuriy Tolmachev
July 15, 2019I see in Electroanalysis current distribution an option Transport Mechanisms/Additional transport mechanisms/Migration in electric field , but I cannot find a detailed description of what this option does. Could you please explain it? Also, is there a comprehensive on-line manual for COMSOL, where we can find answers for such questions by searching a keyword?
Melanie Pfaffe
July 22, 2019Dear Yuriy:
The interface Electroanalysis solves for the transport equation by diffusion. If you also want to consider transport by convection or migration in electric field (electrokinetic flow) you have to enable them in the section transport mechanisms and couple them to a fluid flow interface or electric field interface, respectively.
All the documentation is available with the software. You can use the dynamic help by pressing F1. You will see directly all he information about the Electroanalysis Interface and also crosslinks. In addition we have the documentation available as .pdf via the explorer or within the software.
Pablo Hernández-Arango
September 23, 2020Hello,
in the “Batteries And Fuel Cells Module Users Guide” it is establish that in the molar flux relative to the convective transport is:
Ji = -Di * grad[Ci] – Zi * Um,I * F * Ci *grad[phi]
Then, the definition of the Primary and Secundary current distributions considers cancelling the convection and diffusion terms, it leads to:
il = -F^2 * sum,i(Zi^2 * Um,I * Ci * grad[phi])
Is there not a misunderstanding in the definition of Ji?
Melanie Pfaffe
September 24, 2020 COMSOL EmployeeDear Pablo,
if you first consider the total flux of the species i in the electrolyte Ni, it is the sum over convection (ci*u), diffusion (-Di*grad[Ci]) and migration (-Zi*Um,I *F*Ci*grad[phi]).
Ji is the sum over diffusion and migration without convection: Ji = -Di * grad[Ci] – Zi * Um,I * F * Ci *grad[phi]
This leads to: Ni = Ji +Ci*U
This Ni has to be replaced in the defintion of the current density il = F*sum,i(Zi*Ni), which leads to:
il = F*sum,i(Zi*Ji+Zi*Ci*U)
For electroneutrality with charge conservation the sum of the charges is 0. Therefore the term Zi*Ci*U cancels out.
It remains il = F*sum,i(Zi*Ji)
If you replace Ji you will get: il = -F*sum,i(Zi^2*F*Um,I*Ci*grad[phi])+Zi*Di*grad[Ci])
For perfectly mixed primary and secondary current distribution grad[Ci] = 0
So you end up with: il = -F*sum,i(Zi^2*F*Um,I*Ci*grad[phi]))
which is the same as il = -F^2 * sum,i(Zi^2*Um,I*Ci*grad[phi])
Joseph Benedict
August 4, 2021Fantastic. I like your post and I share my social Networks!
Melanie Pfaffe
August 4, 2021 COMSOL EmployeeThanks a lot Joseph!